
A human blood protein regulates wound healing cells
We tend to think of white blood cells as fighters that attack and kill infections. In the 1850’s, James Paget noticed that some white blood cells can also leave the blood, enter a tissue, and become large, elongated cells now called fibrocytes that help to orchestrate wound healing. Little was known about the signals that tell the white blood cells to become a fibrocyte, or not to become a fibrocyte. We found that a human blood protein called Serum Amyloid P (SAP) prevents the white blood cells from becoming fibrocytes (Figure 1).

Figure 1. When cultured in the presence of serum, human white blood cells remain small, round cells (left panel). In the absence of serum, some human white blood cells become fibrocytes (right panel). Purification of the factor in serum that inhibits fibrocyte differentiation led to the identification of SAP.
A potential therapeutic for wound healing
Wound healing is a major medical problem. Some wounds, such as pressure ulcers, venous ulcers, and diabetic ulcers are very difficult to heal. In the US, there are approximately 85,000 amputations per year because of non-healing diabetic foot ulcers. Because fibrocytes help to form wound-healing tissue, and because SAP inhibits fibrocyte differentiation, we tested the hypothesis that removing SAP from a wound might help healing. Agarose is an edible polymer made from seaweed, and SAP binds very strongly to certain types of agarose in the presence of calcium salts. A wound dressing made with agarose and a small amount of CaCl2 strongly potentiated wound healing in an animal model, and the wounds healed faster than wound treated with a variety of different commercial wound dressings. Patents for this wound dressing have been issued, and this technology is currently available for licensing or partnering.
A potential therapeutic for fibrosis
Like many of us, cells in the body can sometimes overreact to a situation. Cells in the liver, heart, kidneys, lungs, and other tissues can sometimes ‘think’ that they have been wounded. These cells then appear to recruit white blood cells, and promote fibrocyte formation to heal the imaginary wound. Unfortunately, the resulting scar tissue disrupts the structure and function of the tissue, resulting in diseases such as cirrhosis of the liver, heart failure, end-stage kidney disease, pulmonary fibrosis, and other maladies in the 62 or more known fibrosing diseases. Collectively, fibrosing diseases are associated with 30-45% of deaths in the US, and there is no FDA-approved therapy that reverses these diseases. Because fibrocytes participate in fibrosis, and SAP inhibits fibrocyte formation, we tested the hypothesis that injections of SAP might inhibit fibrosis. To our surprise and delight, we found that injections of SAP in an animal model of pulmonary fibrosis blocked the fibrosis. This promoted the formation of a company called Promedior. SAP (also known as pentraxin 2 or PTX2; the recombinant SAP used for clinical trials is called PRM-151) has shown efficacy in 20 different animal models of fibrosing diseases. A 117-patient Phase 2 trial for PRM-151 in pulmonary fibrosis patients showed efficacy better than current of standard of care, and few side effects (see Raghu et al., Effect of Recombinant Human Pentraxin 2 vs Placebo on Change in Forced Vital Capacity in Patients with Idiopathic Pulmonary Fibrosis: A Randomized Clinical Trial. JAMA. 2018 Jun 12;319(22):2299-2307). Roche purchased Promedior, and is now doing the Phase 3 “STARSCAPE” trial for PRM-151 in pulmonary fibrosis patients. See Pilling, D. and Gomer, R.H. The development of serum amyloid P as a possible therapeutic. Frontiers in Immunology, 9, 2328 (2018) for a review of SAP as a therapeutic.
A second-generation therapeutic for fibrosis
We found that a key SAP receptor is DC-SIGN, and that small-molecule ligands of DC-SIGN inhibit fibrocyte differentiation and push macrophages to a Mreg regulatory phenotype. One of the small molecule DC-SIGN ligands inhibits pulmonary fibrosis in a mouse model at the remarkably low dose of 0.001 mg/kg. This technology is currently available for licensing.
What might trigger fibrosis?
In addition to SAP, we found that the protein Slit2 secreted by normal fibroblasts prevents fibrocyte differentiation, and thus appears to be a signal from the fibroblasts in a normal healthy tissue telling the entering monocytes “Everything is fine here, we don’t need any more fibroblasts or fibrocytes”. Conversely, activated fibroblasts secrete the protein lumican, which potentiates fibrocyte differentiation. Since fibrocytes activate fibroblasts, this then forms a positive feedback loop that helps to drive fibrosis. We envision that blocking fibrocyte differentiation with SAP or a DC-SIGN ligand then shuts down this positive feedback loop to stop fibrosis. Other factors that we found to potentiate fibrocyte differentiation and thus possibly initiate fibrosis include certain components of peritoneal dialysis fluid, salt, tryptase released by activated mast cells, and thrombin activated during blood clotting.
Identifying a key driver of fibrosis, and possible third-generation therapeutics for fibrosis
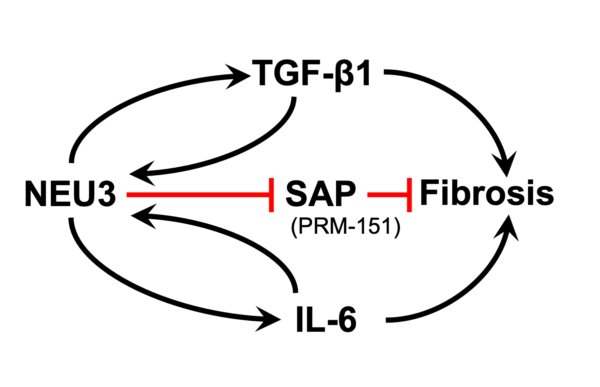
Sialic acid is often the distal sugar on glycoconjugates, and sialidase enzymes remove this sugar. We found extensive desialylation of glycoconjugates in human and mouse fibrotic lesions, and upregulation of the extracellular sialidase NEU3 in fibrotic lesions in idiopathic pulmonary fibrosis and other fibrosing diseases. Mice lacking NEU3 essentially do not develop fibrosis in the bleomycin model, and aspiration of mouse NEU3 (but not enzyme-dead NEU3) causes pulmonary fibrosis in mice. Inhibitors of NEU3 significantly reduce fibrosis in the mouse bleomycin model. Together, this suggests that NEU3 is necessary and sufficient for pulmonary fibrosis in mice. In two positive feedback loops, the profibrotic cytokines TGF-b1 and IL-6 upregulate NEU3 in human lung cells, and NEU3 upregulates these cytokines. For the TGF-b1 loop, NEU3 desialylates a protein that holds extracellular TGF-b1 in an inactive form, causing release of active TGF-b1, and TGF-b1 causes cells to upregulate translation of existing NEU3 mRNA rather than increasing levels of NEU3 mRNA. In collaboration with Tom Meek, we found highly potent transition state analogue inhibitors of NEU3, and showed that these are potent anti-fibrotics in a mouse model of pulmonary fibrosis. This technology is currently available for licensing.
Key papers
Pilling, D., Buckley, C.D., Salmon, M., and Gomer, R.H. Inhibition of fibrocyte differentiation by serum amyloid P. J. Immunology 171, 5537-5546 (2003).
Pilling, D., Tucker, N., and Gomer, R.H. Aggregated IgG inhibits human fibrocyte differentiation. J. Leukocyte Biology, 79, 1242-1251 (2006).
Haudek, S.B., Xia, Y., Huebener, P., Lee, J.M., Carlson, S., Crawford, J.R., Pilling, D., Gomer, R.H., Trial, J., Frangogiannis, N.G., and Entman, M.L. Bone Marrow-derived Fibroblast Precursors Mediate Ischemic Cardiomyopathy in Mice. Proc. Natl. Acad. Sci. USA, 103, 18284-18289 (2006).
Pilling, D., Roife, D., Wang, M., Ronkainen, S.D., Crawford, J.R., Travis, E.L., and Gomer, R.H. Reduction of bleomycin-induced pulmonary fibrosis by serum amyloid P. J. Immunology, 179, 4035-4044 (2007).
Naik-Mathuria, B., Pilling, D., Crawford, J.R., Gay, A.N., Smith, C.W., Gomer, R.H., and Olutoye, O.O. Serum Amyloid P inhibits dermal wound healing. Wound Repair and Regeneration, 16, 266-273 (2008).
Shao, D.D., , R., Vakil, V., Gomer, R.H., and Pilling, D. Th-1 cytokines inhibit, and Th-2 cytokines promote fibrocyte differentiation. Journal of Leukocyte Biology. 83, 1323-1333 (2008).
Gomer, R.H., Pilling, D, Kauvar, L.M., Ellsworth, S., Ronkainen, S.D., Roife, D., and Davis, S.C. A Serum Amyloid P-binding hydrogel speeds healing of partial thickness wounds in pigs. Wound Repair and Regeneration, 17, 397-404 (2009).
Pilling, D., Fan, T., Huang, D., Kaul, B., and Gomer, R.H. Identification of markers that distinguish monocyte-derived fibrocytes from monocytes, macrophages, and fibroblasts. PLoS ONE, 4, e7475 (2009).
Crawford, J.R., Bjorklund, N.L., Taglialatela, G., and Gomer, R.H. Brain Serum amyloid P levels are reduced in individuals that lack dementia while having Alzheimer’s disease neuropathology. Neurochemical Research, 37, 795-801 (2012).
Crawford, J.R., Pilling, D., and Gomer, R.H. FcγRI mediates serum amyloid P inhibition of fibrocyte differentiation. Journal of Leukocyte Biology, 92, 699-711 (2012).
Cox, N., Pilling, D., and Gomer, R.H. NaCl potentiates human fibrocyte differentiation. PLoS ONE, 7, e45674 (2012).
Maharjan, A.S., Roife, D., Brazill, D., and Gomer, R.H. Serum Amyloid P inhibits granulocyte adhesion. Fibrogenesis & Tissue Repair, 6, 2 (2013).
White, M.J.V., Glenn, M., and Gomer, R.H. Trypsin potentiates human fibrocyte differentiation. PLoS ONE, 8, e70795 (2013).
Gomer, R.H. New approaches to modulating idiopathic pulmonary fibrosis. Current Allergy and Asthma Reports, 13, 607-612 (2013).
Pilling, D. and Gomer, R.H. Persistent lung inflammation and fibrosis in Serum Amyloid P (Apcs-/-) knockout mice. PLoS ONE, 9, e93730 (2014).
Pilling, D., Crawford, J.R., Verbeek, J.S., and Gomer, R.H. Inhibition of murine fibrocyte differentiation by cross-linked IgG is dependent on FcgRI. Journal of Leukocyte Biology, 96, 275-282 (2014).
Cox, N., Pilling, D., and Gomer, R.H. Distinct Fcγ receptors mediate the effect of Serum Amyloid P on neutrophil adhesion and fibrocyte differentiation. Journal of Immunology, 193, 1701-8 (2014).
Cox, N., Pilling, D., and Gomer, R.H. Serum Amyloid P: a systemic regulator of the innate immune response. Journal of Leukocyte Biology, 96, 739-743 (2014).
Pilling, D., Zheng, Z., Vakil, V., and Gomer, R.H. Fibroblasts secrete Slit2 to inhibit fibrocyte differentiation and fibrosis. Proc. Natl. Acad. Sci. USA, 111, 18291-18296 (2014).
White, M.J.V., Galvis-Carvajal, E., and Gomer, R.H. A brief exposure to tryptase or thrombin potentiates fibrocyte differentiation in the presence of serum or serum amyloid P. Journal of Immunology, 194, 142-150 (2015).
Pilling, D., Cox, N., Vakil, V., Verbeek, J.S., and Gomer, R.H. The long pentraxin PTX3 promotes fibrocyte differentiation. PLoS ONE, 10, e0119709 (2015).
Cox, N., Pilling, D., and Gomer, R.H. DC-SIGN activation mediates the differential effects of SAP and CRP on the innate immune system and inhibits fibrosis in mice. Proc. Natl. Acad. Sci. USA, 112, 8385-8390 (2015).
White, M.J.V., Roife, D., and Gomer, R.H. Galectin-3 binding protein secreted by breast cancer cells inhibits monocyte-derived fibrocyte differentiation. Journal of Immunology, 195, 1858-1867 (2015).
Pilling, D., Vakil, V., Cox, N., and Gomer, R.H. TNF-a-stimulated fibroblasts secrete lumican to promote fibrocyte differentiation. Proc. Natl. Acad. Sci. USA, 112, 11929-11934 (2015).
White, M.J.V. and Gomer, R.H. Trypsin, tryptase, and thrombin polarize macrophages towards a pro-fibrotic M2a phenotype. PLoS ONE, 10, e0138748 (2015).
Herlihy, S.E., Stark, H.E., Lopez-Anton, M., Cox, N., Keyhanian, K., Fraser, D.J., and Gomer, R.H. Peritoneal dialysis fluid and some of its components potentiate fibrocyte differentiation. Peritoneal Dialysis International, 36, 367-373 (2016).
Pilling, D., Galvis-Carvajal, E., Karhadkar, T., Cox, N., and Gomer, R.H. Monocyte differentiation and macrophage priming are regulated differentially by pentraxins and their ligands. BMC Immunology, 18, 30 (2017).
Karhadkar, T.R., Pilling, D., Cox, N., and Gomer, R.H. Sialidase inhibitors attenuate pulmonary fibrosis in a mouse model. Scientific Reports, 7, 15069 (2017).
Chen, W., Pilling, D., and Gomer, R.H. Dietary NaCl affects bleomycin-induced lung fibrosis in mice. Experimental Lung Research, 43, 395-406 (2017).
Pilling, D. and Gomer, R.H. The development of serum amyloid P as a possible therapeutic. Frontiers in Immunology, 9, 2328 (2018).
Pilling, D., Cox, N., Thompson, M.A., Karhadkar, T.R., and Gomer, R.H. Serum amyloid P and a Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Nonintegrin ligand inhibit high fat diet-induced adipose tissue and liver inflammation and steatosis in mice. American Journal of Pathology, 189, 2400-2413 (2019).
Karhadkar, T.R., Chen, W., and Gomer, R.H. Attenuated pulmonary fibrosis in sialidase-3 knockout (Neu3-/-) mice. American Journal of Physiology-Lung Cellular and Molecular Physiology, 318, L165-L179 (2020).
Chen, W., Lamb, T.R., and Gomer, R.H. TGF-β1 Increases Sialidase 3 Expression in Human Lung Epithelial Cells by Decreasing its Degradation and Upregulating its Translation. Experimental Lung Research, 46, 75-80 (2020).
Chen, W., Karhadkar, T.R., Ryu, C., Herzog, E.L., and Gomer, R.H. Reduced sialylation and bioactivity of the anti-fibrotic protein serum amyloid P in the sera of patients with idiopathic pulmonary fibrosis. ImmunoHorizons, 4, 352-362 (2020).
Pilling, D., Karhadkar, T.R., and Gomer, R.H. A CD209 ligand and a sialidase inhibitor differentially modulate adipose tissue and liver macrophage populations and steatosis in mice on the methionine and choline-deficient (MCD) diet. PLoS ONE, 15, e0244762 (2020).
Pilling, D., Karhadkar, T.R., and Gomer, R.H. High-fat diet-induced adipose tissue and liver inflammation and steatosis in mice are reduced by inhibiting sialidases. American Journal of Pathology, 191, 131-143 (2021).
Karhadkar, T.R., Meek, T.D., and Gomer, R.H. Inhibiting sialidase-induced TGF-β1 activation attenuates pulmonary fibrosis in mice. Journal of Pharmacology and Experimental Therapeutics, 376, 106-117 (2021).
Karhadkar, T.R., Pilling, D., and Gomer, R.H. Serum Amyloid P inhibits single stranded RNA-induced lung inflammation, lung damage, and cytokine storm in mice. PLoS ONE, 16, e0245924 (2021).